Covid-19
22-08-2024
Publication date: 07-08-2021
Updated on: 16-05-2025
Topic: Covid-19
Estimated reading time: 1 min
Article Author
Eufemia PutortiMedical Editor
Massimo Degano
Editor and Translator
Viktoryia LuhakovaThe ongoing COVID-19 pandemic has forced the scientific community to devote itself to the research and development of various strategies to fight SARS-CoV-2 infection. The only informed way to identify new antivirals is through the knowledge of the three-dimensional structure, literally the shape, of the virus proteins that are responsible for cell infection and virus replication. Once the structure of proteins is known, it is possible for chemists to synthesize compounds that block its function, and consequently the virus life cycle.
We talk about it in this cycle of 5 episodes with the help of Dr. Massimo Degano, Group leader of the Biocrystallography Unit of the San Raffaele Hospital and lecturer at our University.
We explained in the previous episodes that, in order to develop drugs against SARS-CoV-2, it is necessary to know with high detail (resolution) the structure of the proteins of the virus that we have chosen as targets based on their importance for the life cycle of the virus. This is the task of structural biology, and we have already explained the principle of operation of cryo-electron microscopy. The first technique that allowed to visualize the structures of molecules with atomic detail, and still provides a very high detail, is X-ray crystallography.
The crystallographic theory was developed shortly after Wilhelm Röntgen discovered X-rays, thanks to contributions from Max von Laue (Nobel prize winner in 1914) and William Henry and William Lawrence Bragg (father and son, both awarded Nobel prize 1915), and over the years it has been constantly refined and improved. At the time of these pioneers determining the structure of small molecules such as aspirin was a formidable challenge, but thanks to visionaries such as Sir John Kendrew and Max Perutz it was possible to determine the first structures of proteins, myoglobin and hemoglobin. From 1945 to today, the list of successes in crystallography in providing us with images of proteins of biomedical interest has been infinite, and the importance of this information is corroborated by the Royal Swedish Academy of Sciences which regularly rewards the structural studies that allow us with the Nobel Prize to understand the functioning of complex proteins (https://www.iucr.org/people/nobel-prize).
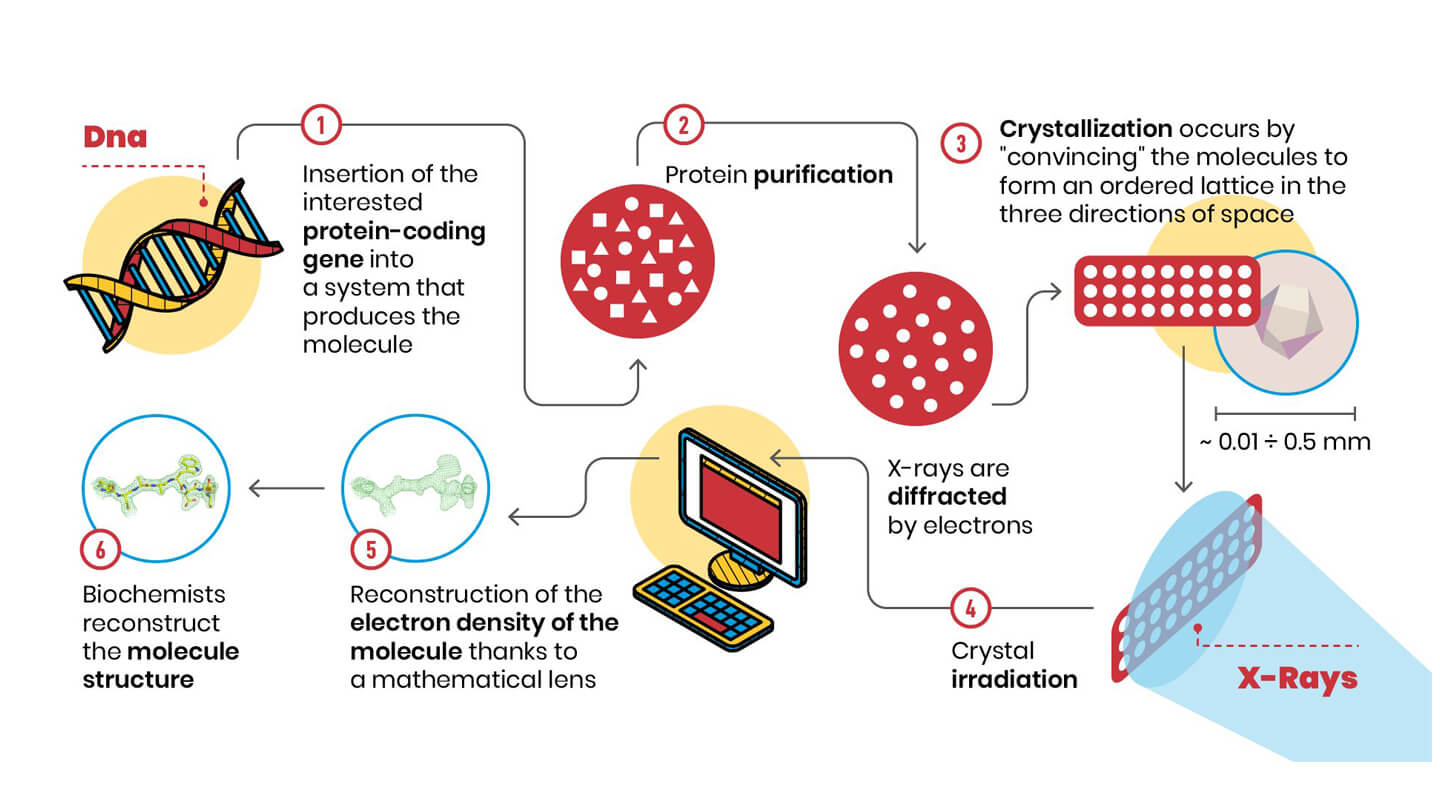
What are the steps of this technique? Let’s look at the scheme:
1. 2. First of all, it is necessary to have milligrams of pure sample available, that is, infinitesimal quantities of contaminants: in practice, many identical molecules.
3. Next, these molecules must form a crystal, i.e. an ordered three-dimensional lattice. This step is fundamental, because the molecules in the crystal are positioned according to symmetry patterns, such as those studied by M. C. Escher (https://mcescher.com/gallery/symmetry/).
4. The order of the molecules in the crystal causes the electrons around the atoms, when irradiated with X-rays, to scatter the radiation in phase, adding up just like the components of a vocal choir. This amplification effect allows us to measure the diffraction spectrum, i.e. the positions and how intense the X-rays diffracted by the molecules in the crystal are.
5. Once this measurement is made, and by applying a sort of mathematical lens (called “the inverse Fourier transform”), we obtain the electron density map that tells us how the atoms are arranged in the crystal. The electron density can be viewed on the computer, and the components of the biological molecule we are studying (amino acids for proteins, ribose and nitrogen bases for DNA and RNA...) must be inserted into it, in an interlocking process that might resemble some video games.
6. Having completed the interpretation of electronic density, we finally have the three-dimensional structure!
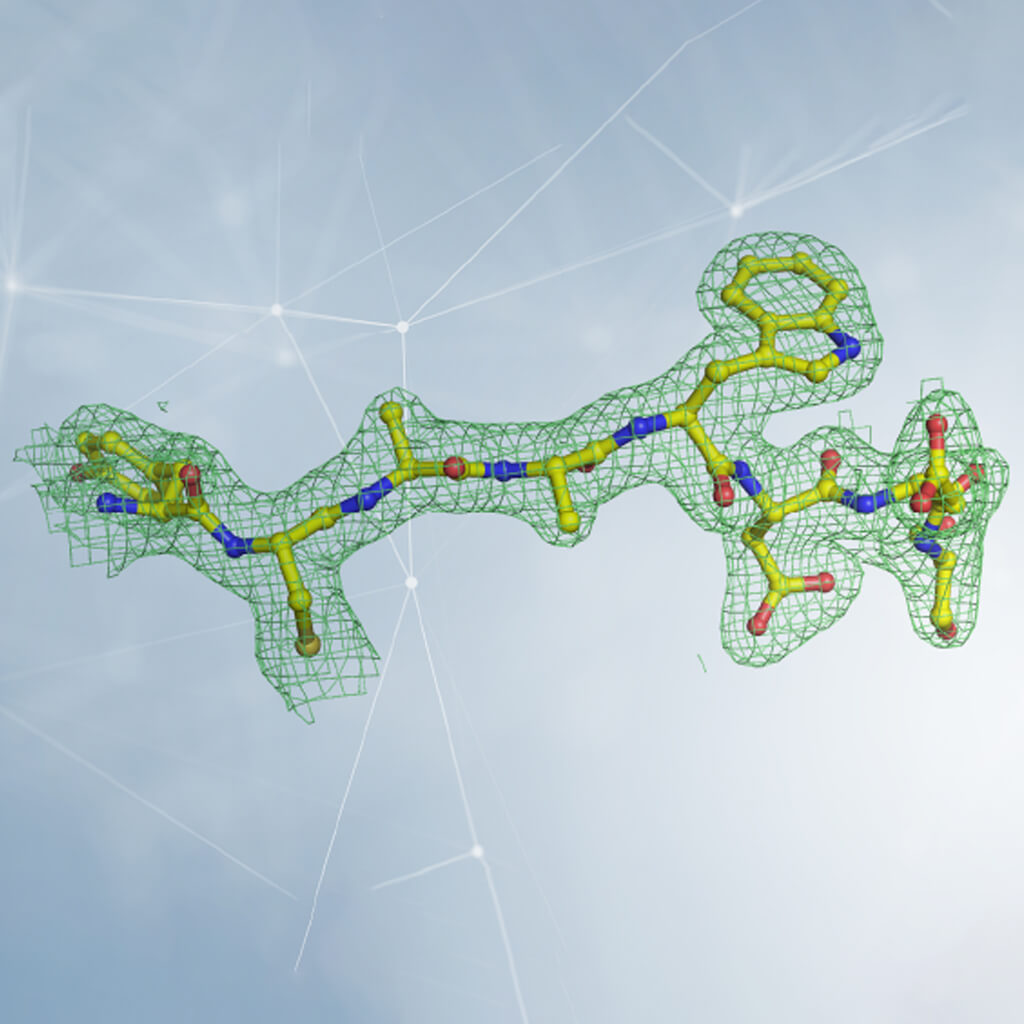
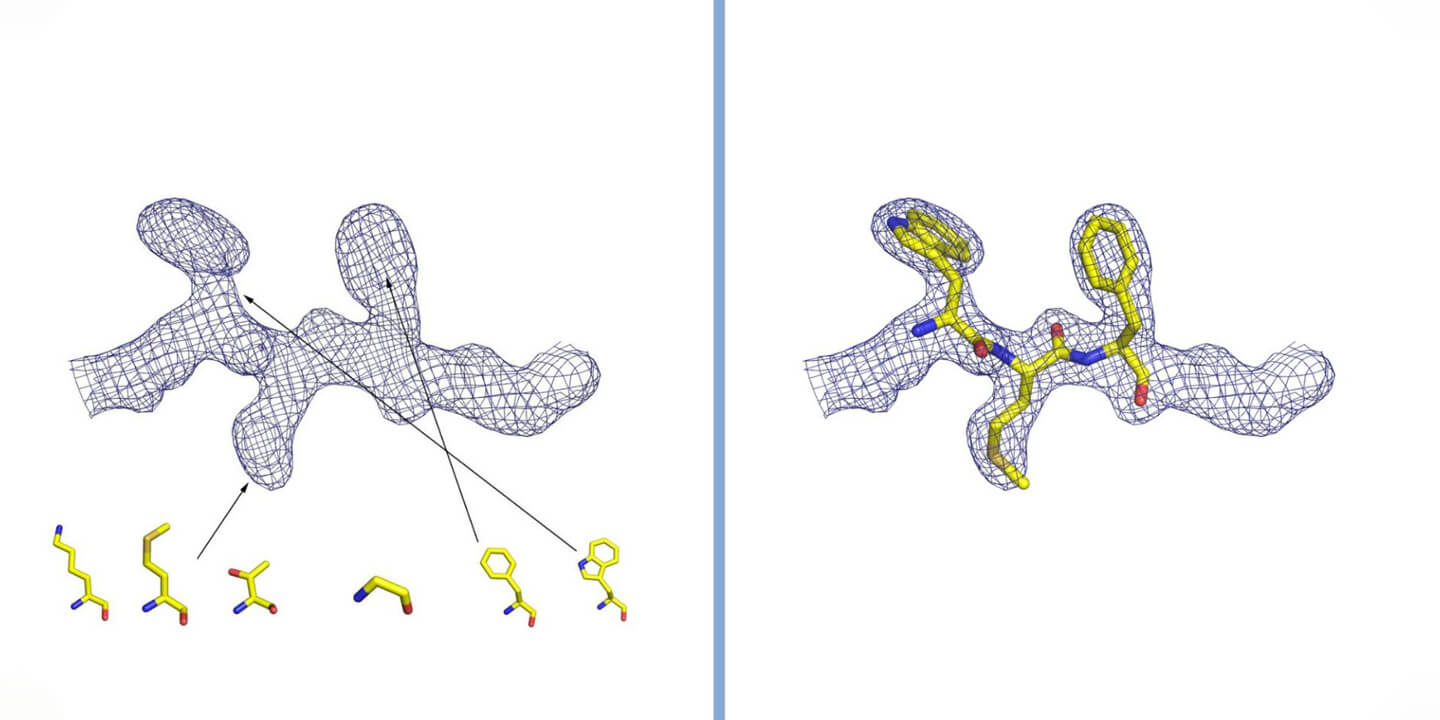
Interpretation of electron density in crystallography. The density is represented by a grid, inside which are the atoms that make up the protein. In the case of a protein, the shape of the density is observed and compared with the structure of the twenty amino acids that constitute it (some of which are represented in the figure). By inserting the amino acids in the correct order and following the three-dimensional winding of the electron density, the structure of the protein is reconstructed. Courtesy of Dr. Degano.
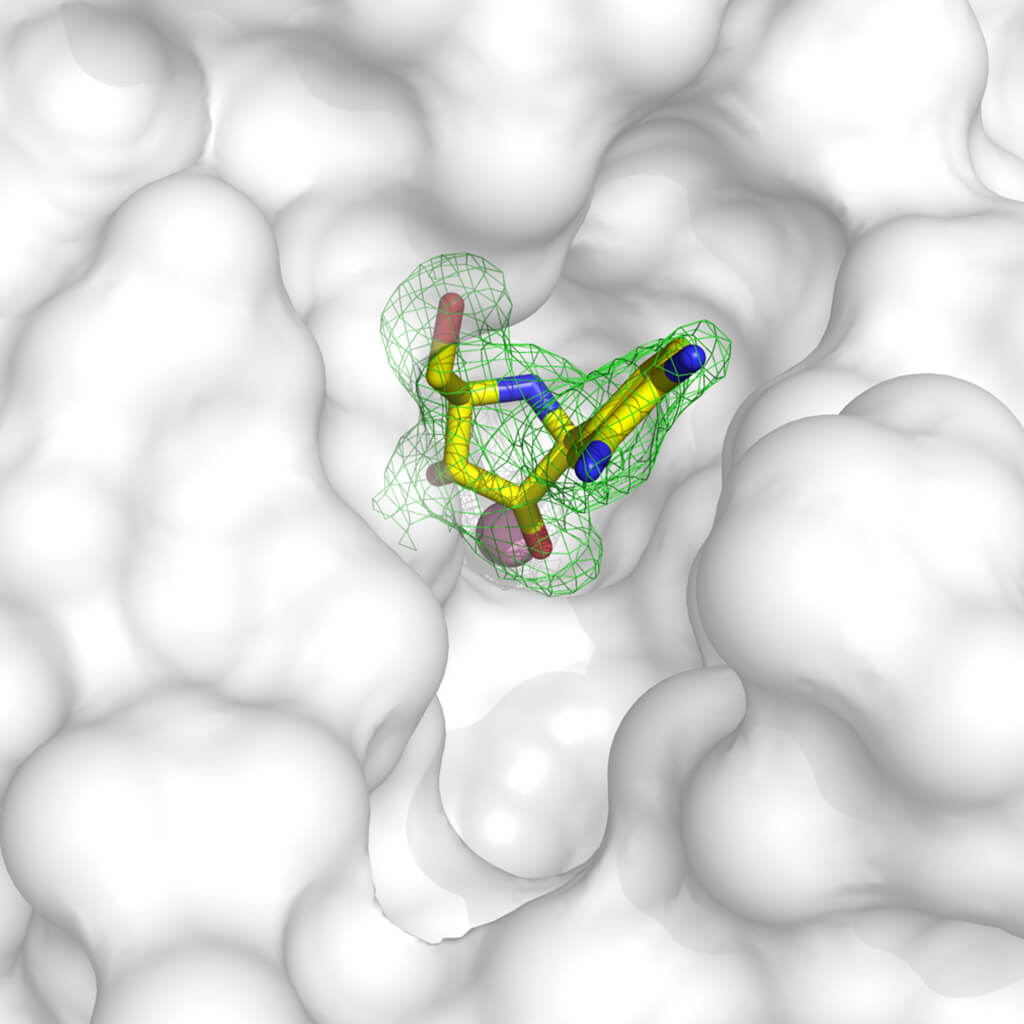
Crystallography is a very versatile and powerful tool for the development of lead compounds with the prospect of becoming drugs. With various methodologies, it is possible to obtain the structure of target proteins linked to molecules with inhibitory activity: in fact, it is possible to “soak” the crystals in a solution in which a substance that we know is dissolved can bind to the target protein, or obtain protein crystals after having it tied to the compound. Once the structure of the protein bound to a compound is known, we can visualize which parts of the protein (which we can assimilate to a glove) are not correctly filled by the molecule (the fingers of a hand). With this information we can modify the inhibitor molecule to make it better complement our target, and block its function!
Example of inhibitor of an enzyme that has been inserted into the molecules of a crystal. The electron density in green clearly defines where the molecule is positioned in the protein structure, represented with the white surface. It can be seen that the molecule does not perfectly occupy the available cavity, so it is possible to engineer a compound that better complements the protein structure and efficiently blocks its function. The structure used is accessible with the 3MKN code in the Protein Data Bank. Courtesy of Dr. Degano.