Covid-19
22-08-2024
Fecha de publicación: 07-08-2021
Actualizado en: 16-05-2025
Asunto: Covid-19
Tiempo estimado de lectura: 1 min
Autor del Artículo
Eufemia PutortiEditor médico
Massimo Degano
Editor y Traductor
Viktoryia LuhakovaLa actual pandemia de COVID-19 ha obligado a la comunidad científica a dedicarse a la investigación y el desarrollo de diversas estrategias para combatir la infección por SARS-CoV-2. La única forma informada de identificar nuevos antivirales es mediante el conocimiento de la estructura tridimensional, literalmente la forma, de las proteínas del virus que son responsables de la infección celular y la replicación del virus. Una vez que se conoce la estructura de las proteínas, los químicos pueden sintetizar compuestos que bloqueen su función y, en consecuencia, el ciclo vital del virus.
De ello hablamos en este ciclo de 5 episodios con la ayuda del Dr. Massimo Degano, jefe de grupo de la Unidad de Biocristalografía del Hospital IRCCS San Raffaele y profesor de nuestra Universidad.
Ya explicamos en los episodios anteriores que, para desarrollar fármacos contra el SARS-CoV-2, es necesario conocer con gran detalle (resolución) la estructura de las proteínas del virus que hemos elegido como diana en función de su importancia para el ciclo vital del virus. Esta es la tarea de la biología estructural, y ya hemos explicado el principio de funcionamiento de la criomicrografía electrónica. La primera técnica que permitió visualizar las estructuras de las moléculas con detalle atómico, y que todavía proporciona un detalle muy elevado, es la cristalografía de rayos X.
La teoría cristalográfica se desarrolló poco después de que Wilhelm Röntgen descubriera los rayos X, gracias a las aportaciones de Max von Laue (premio Nobel en 1914) y William Henry con William Lawrence Bragg (padre e hijo, ambos galardonados con el premio Nobel en 1915), y a lo largo de los años se ha ido perfeccionando y mejorando constantemente. En la época de estos pioneros, determinar la estructura de pequeñas moléculas como la aspirina era un reto formidable, pero gracias a visionarios como Sir John Kendrew y Max Perutz fue posible determinar las primeras estructuras de las proteínas, la mioglobina y la hemoglobina. Desde 1945 hasta hoy, la lista de éxitos de la cristalografía a la hora de proporcionarnos imágenes de proteínas de interés biomédico ha sido infinita, y la importancia de esta información la corrobora la Real Academia Sueca de las Ciencias, que premia regularmente con el Nobel los estudios estructurales que permiten comprender el funcionamiento de las proteínas complejas (https://www.iucr.org/people/nobel-prize).
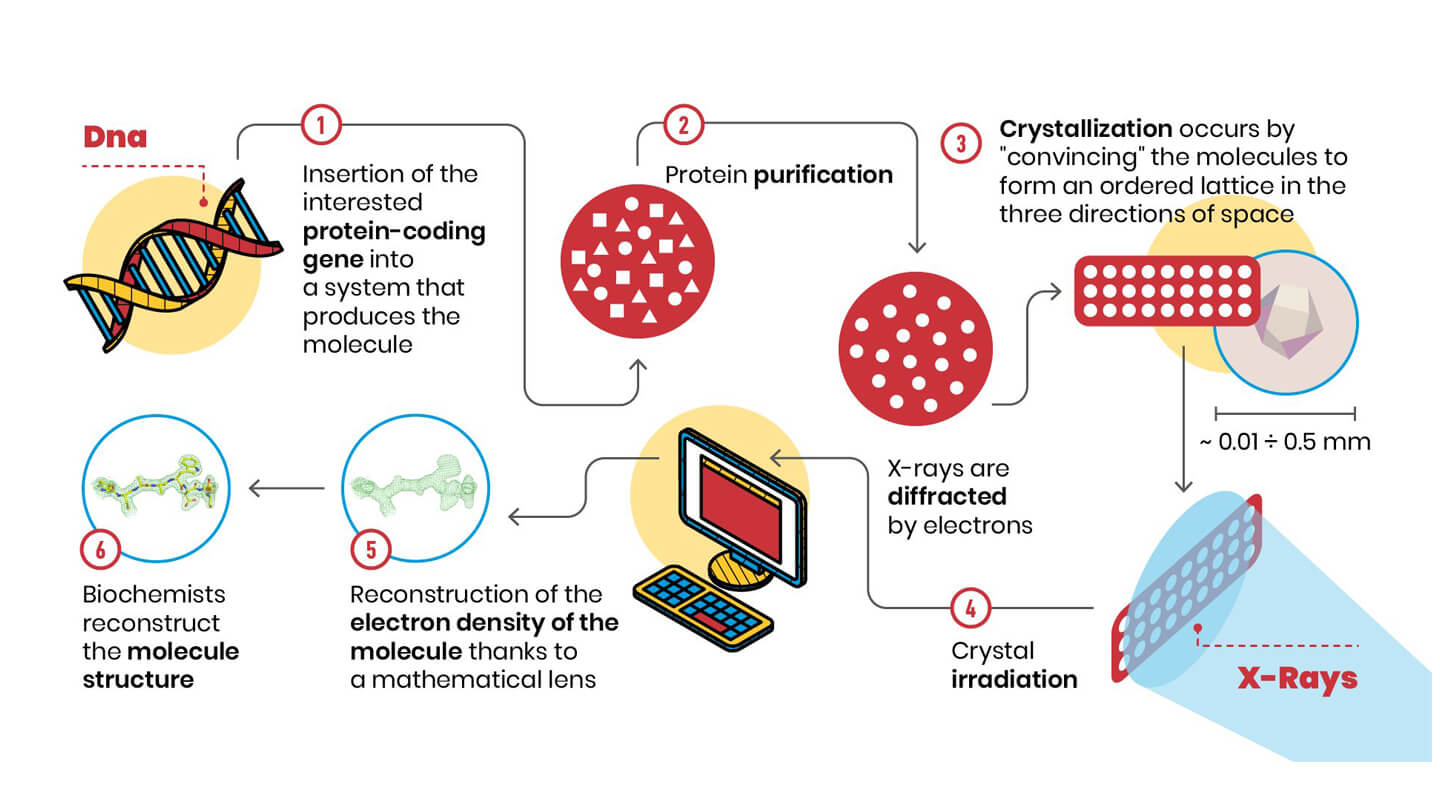
¿Cuáles son los pasos de esta técnica? Veamos el esquema:
1. 2. En primer lugar, es necesario disponer de miligramos de muestra pura, es decir, de cantidades infinitesimales de contaminantes: en la práctica, muchas moléculas idénticas.
3. A continuación, estas moléculas deben formar un cristal, es decir, una red tridimensional ordenada. Esta etapa es fundamental, ya que las moléculas del cristal se colocan según patrones de simetría, como los estudiados por M. C. Escher (https://mcescher.com/gallery/symmetry/ ).
4. El orden de las moléculas en el cristal hace que los electrones que rodean a los átomos, al ser irradiados con rayos X, dispersen la radiación en fase, sumándose como los componentes de un coro vocal. Este efecto de amplificación permite medir el espectro de difracción, es decir, las posiciones y la intensidad de los rayos X difractados por las moléculas del cristal.
5. Una vez realizada esta medición, y aplicando una especie de lente matemática (llamada "la transformada inversa de Fourier"), obtenemos el mapa de densidad de electrones que nos indica cómo están dispuestos los átomos en el cristal. La densidad de electrones se puede visualizar en el ordenador, y en ella se deben insertar los componentes de la molécula biológica que estamos estudiando (aminoácidos para las proteínas, bases de ribosa y nitrógeno para el ADN y el ARN...), en un proceso de enclavamiento que podría parecerse a algunos videojuegos.
6. Una vez completada la interpretación de la densidad electrónica, ¡por fin tenemos la estructura tridimensional!
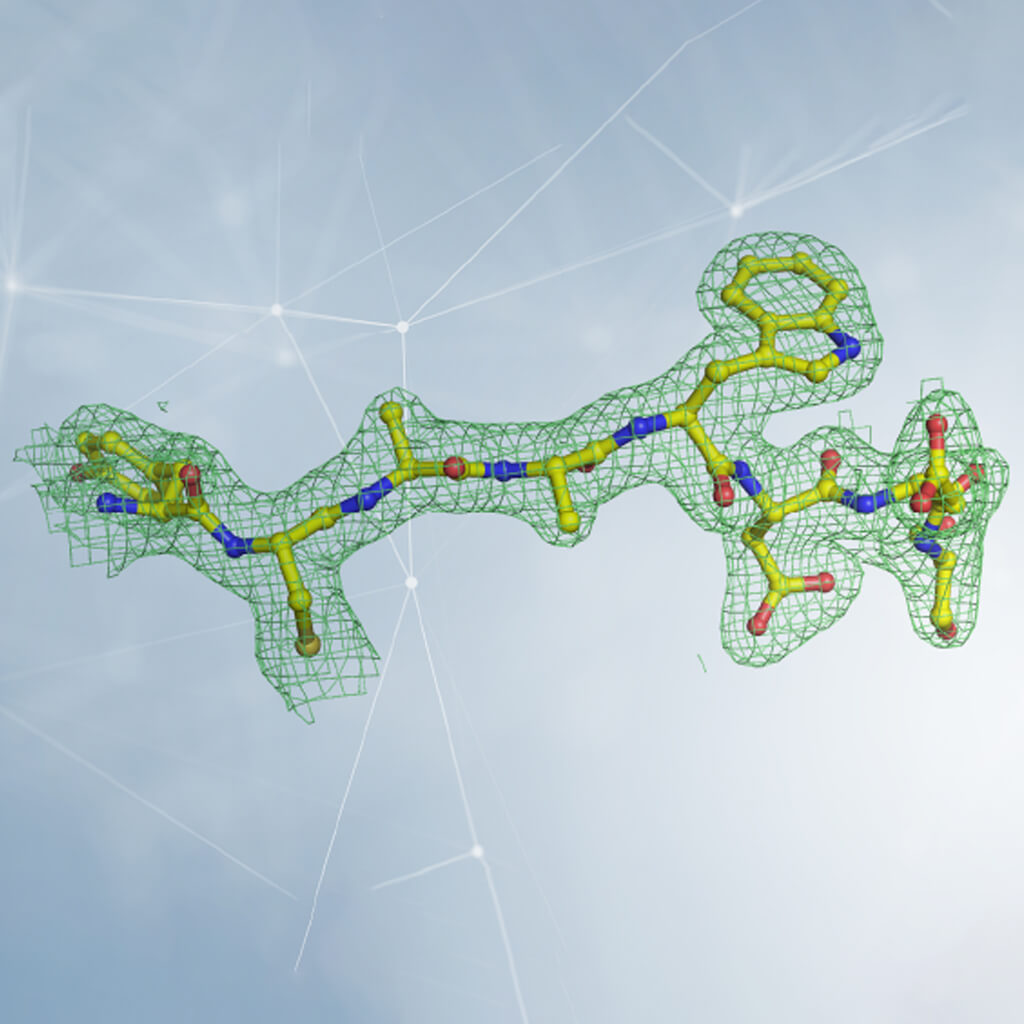
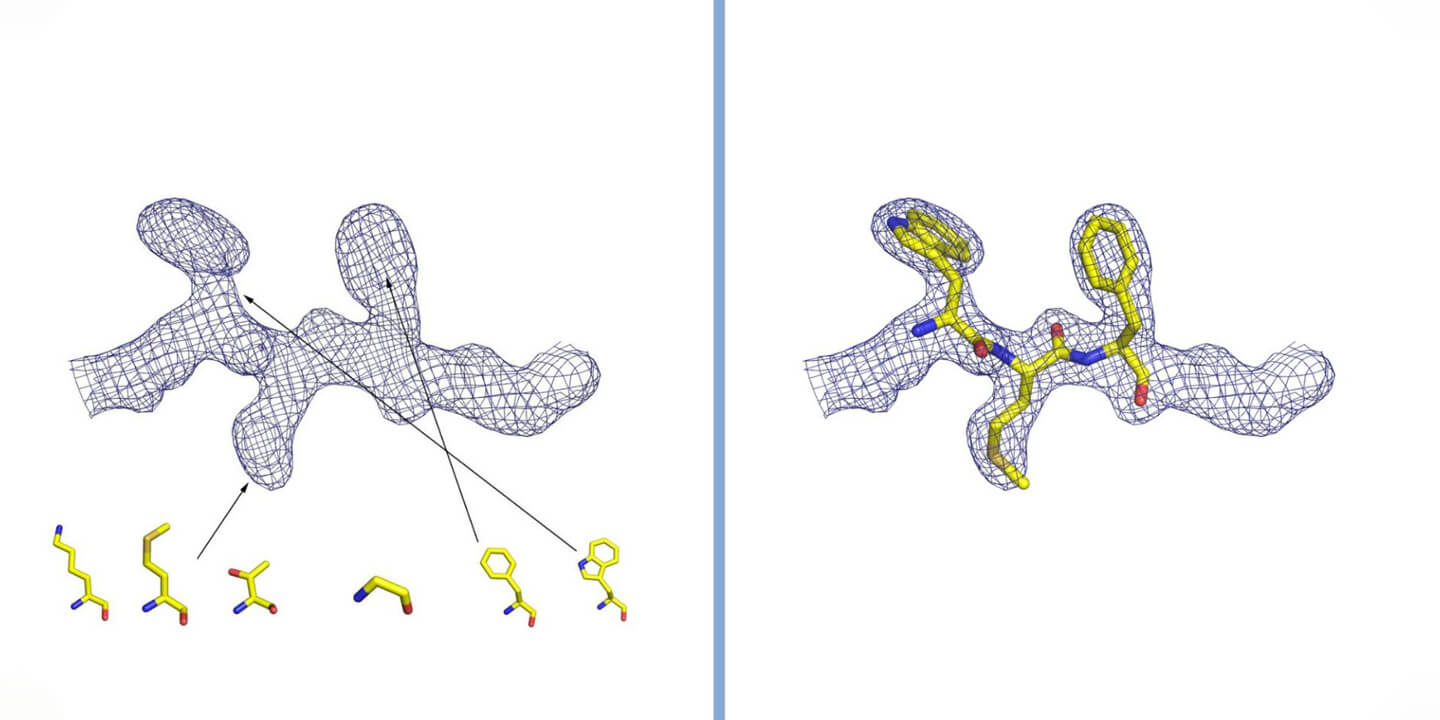
Interpretación de la densidad electrónica en la cristalografía. La densidad está representada por una cuadrícula, dentro de la cual están los átomos que componen la proteína. En el caso de una proteína, se observa la forma de la densidad y se compara con la estructura de los veinte aminoácidos que la constituyen (algunos de los cuales están representados en la figura). Insertando los aminoácidos en el orden correcto y siguiendo la sinuosidad tridimensional de la densidad electrónica, se reconstruye la estructura de la proteína. Cortesía del Dr. Degano.
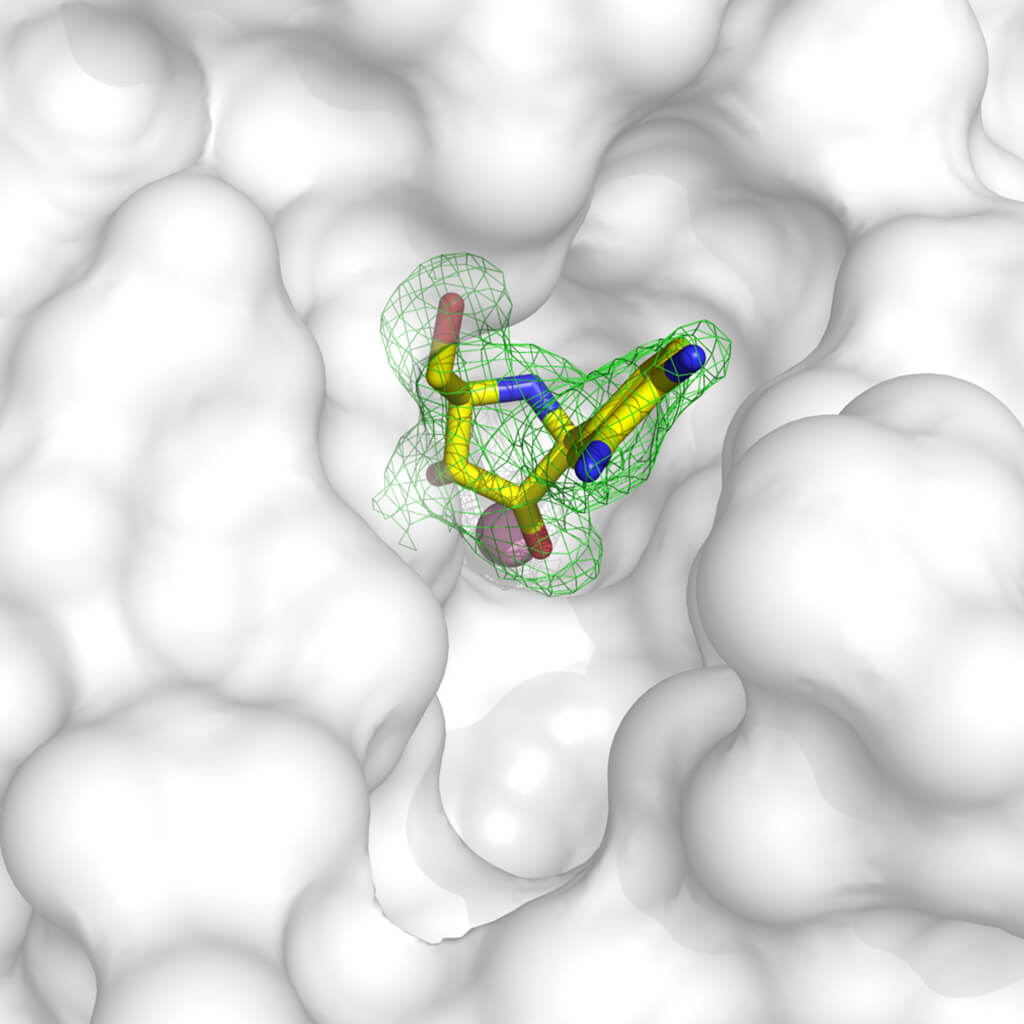
La cristalografía es una herramienta muy versátil y poderosa para el desarrollo de compuestos líderes con perspectivas de convertirse en fármacos. Con diversas metodologías, es posible obtener la estructura de proteínas diana unidas a moléculas con actividad inhibidora: de hecho, es posible "empapar" los cristales en una solución en la que una sustancia que sabemos que está disuelta puede unirse a la proteína diana, u obtener cristales de proteína después de haberla unido al compuesto. Una vez conocida la estructura de la proteína unida a un compuesto, podemos visualizar qué partes de la proteína (que podemos asimilar a un guante) no están correctamente ocupadas por la molécula (los dedos de una mano). Con esta información podemos modificar la molécula inhibidora para que se complemente mejor con nuestra diana, ¡y bloquear su función!
Ejemplo de inhibidor de una enzima que se ha insertado en las moléculas de un cristal. La densidad de electrones en verde define claramente dónde se sitúa la molécula en la estructura de la proteína, representada con la superficie blanca. Se puede ver que la molécula no ocupa perfectamente la cavidad disponible, por lo que es posible diseñar un compuesto que se complemente mejor con la estructura de la proteína y bloquee eficazmente su función. La estructura utilizada es accesible con el código 3MKN en el Protein Data Bank. Cortesía del Dr. Degano.